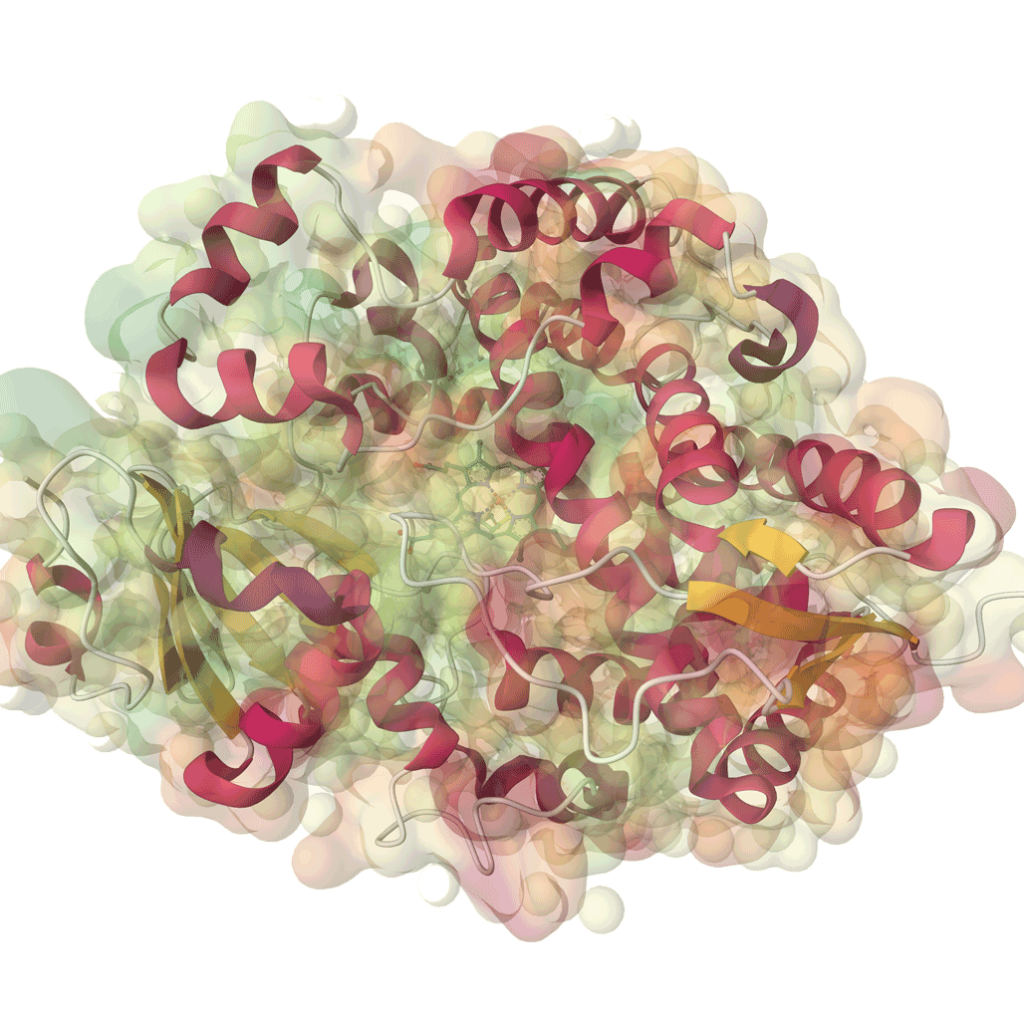
What would be the advantage of using a molecular model? This question is fundamental in the field of material science, where the ability to accurately simulate and predict material behavior can significantly impact research and development. Molecular models allow us to visualize complex interactions at the atomic level, enabling us to design materials with enhanced properties and performance. By leveraging these models, researchers can make informed decisions, streamline their processes, and accelerate innovation.
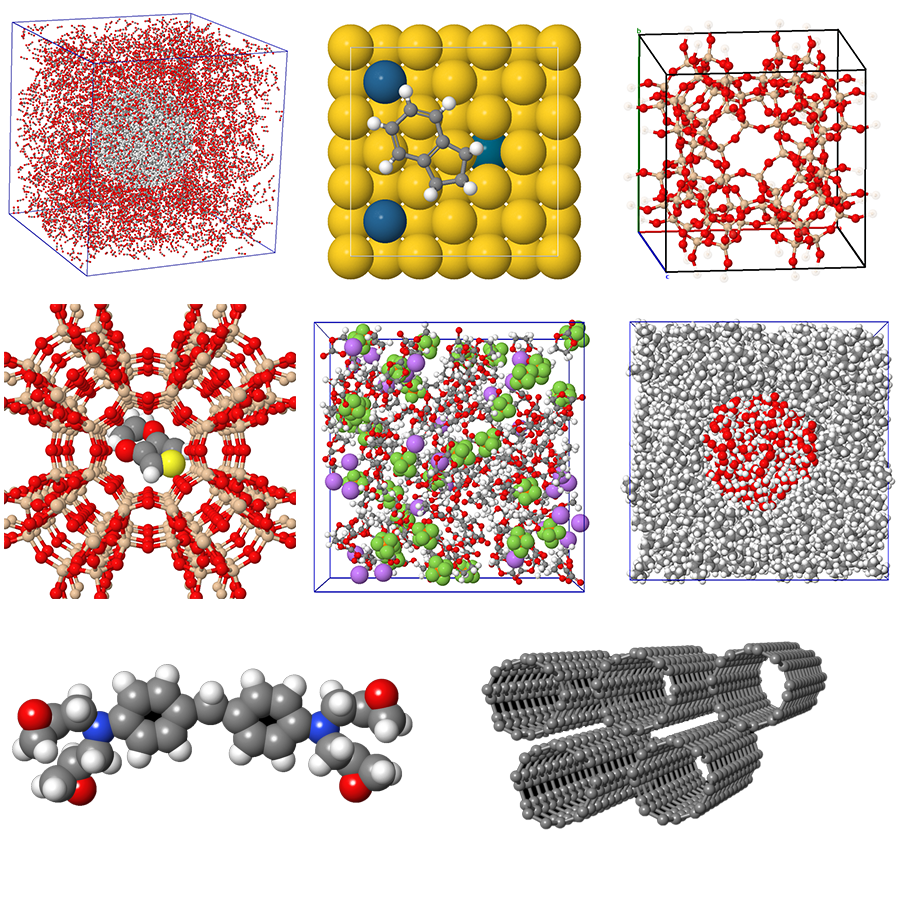
Introducing MaXFlow in Material Science
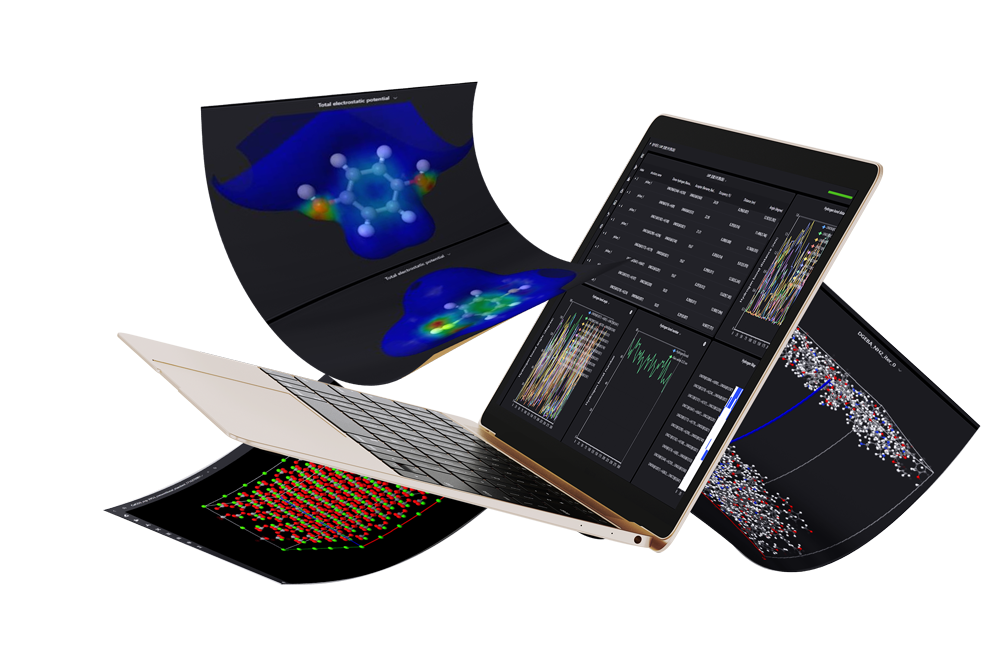
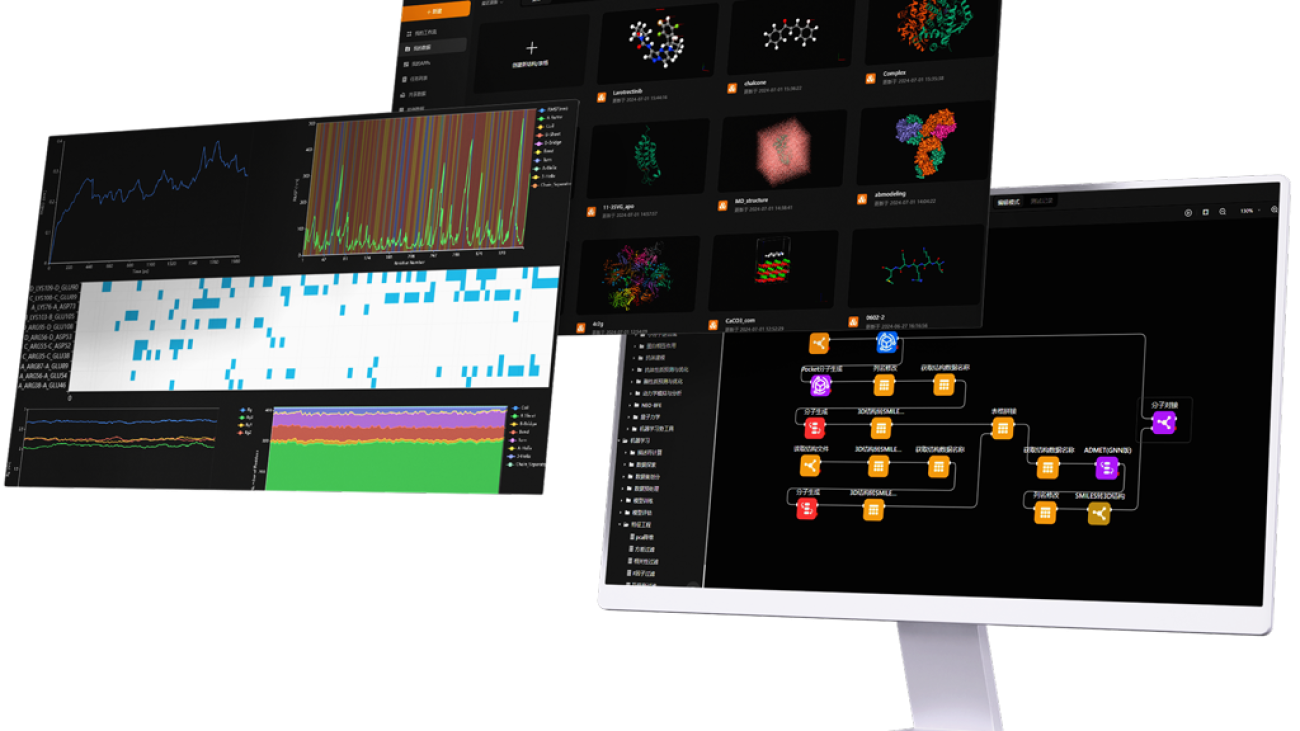
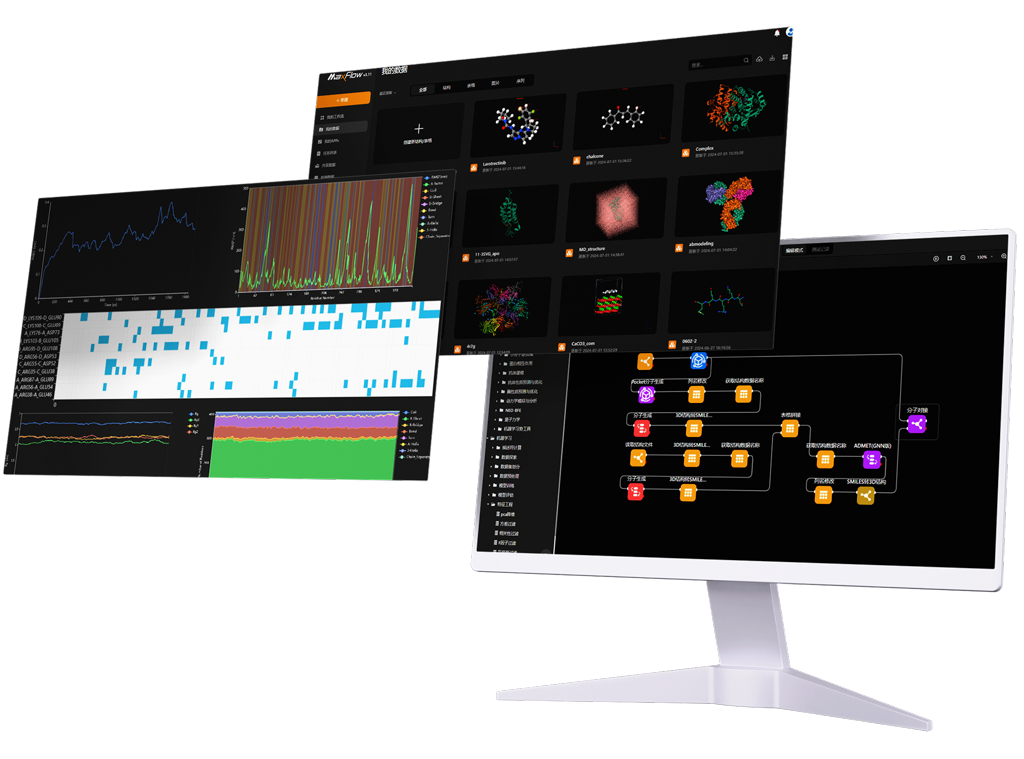
MaXFlow is a cutting-edge molecular simulation and artificial intelligence platform that transforms how we approach materials modeling. By harnessing the power of cloud computing and AI, MaXFlow offers advanced tools for predicting material properties and designing experiments effectively. As we explore what would be the advantage of using a molecular model, it’s clear that MaXFlow empowers researchers to enhance R&D efficiency and drive innovation in materials design.
Innovative Microstructure Design
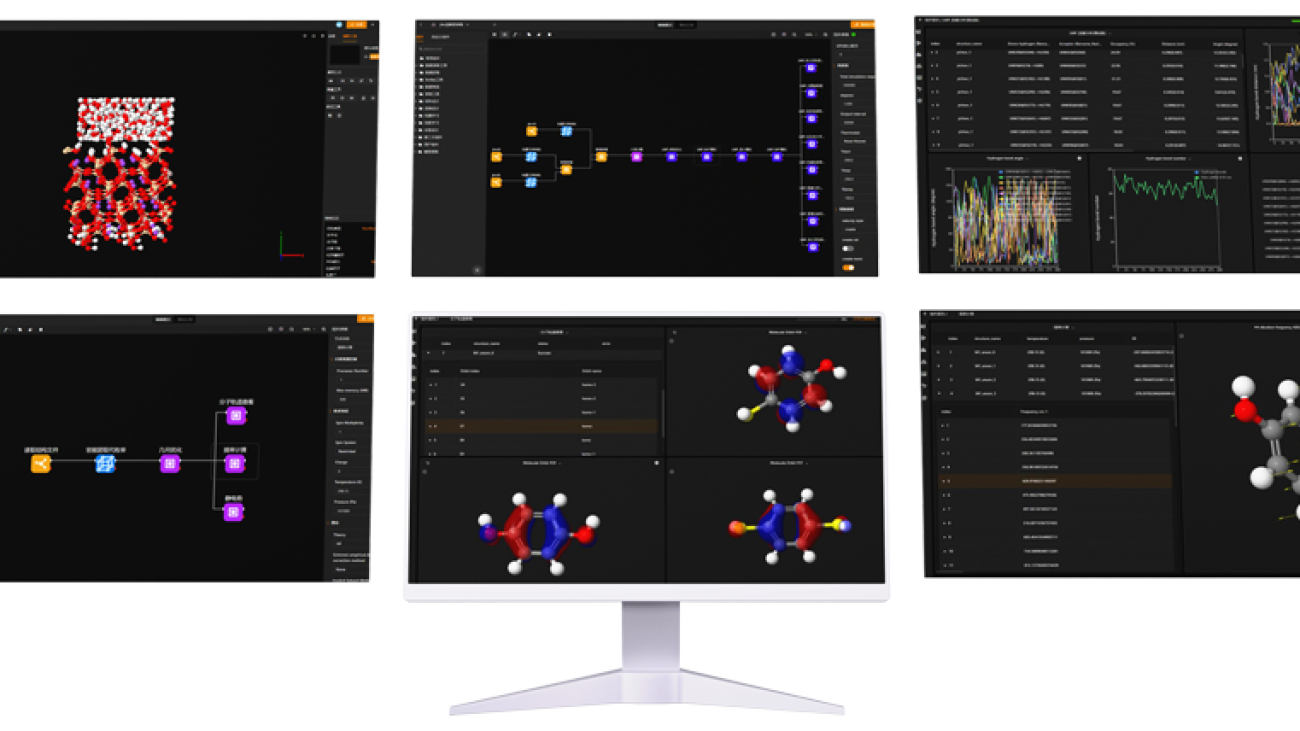
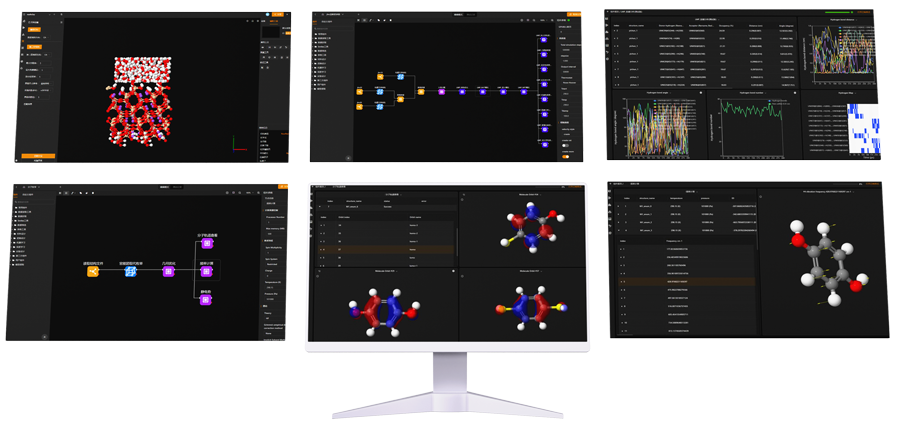
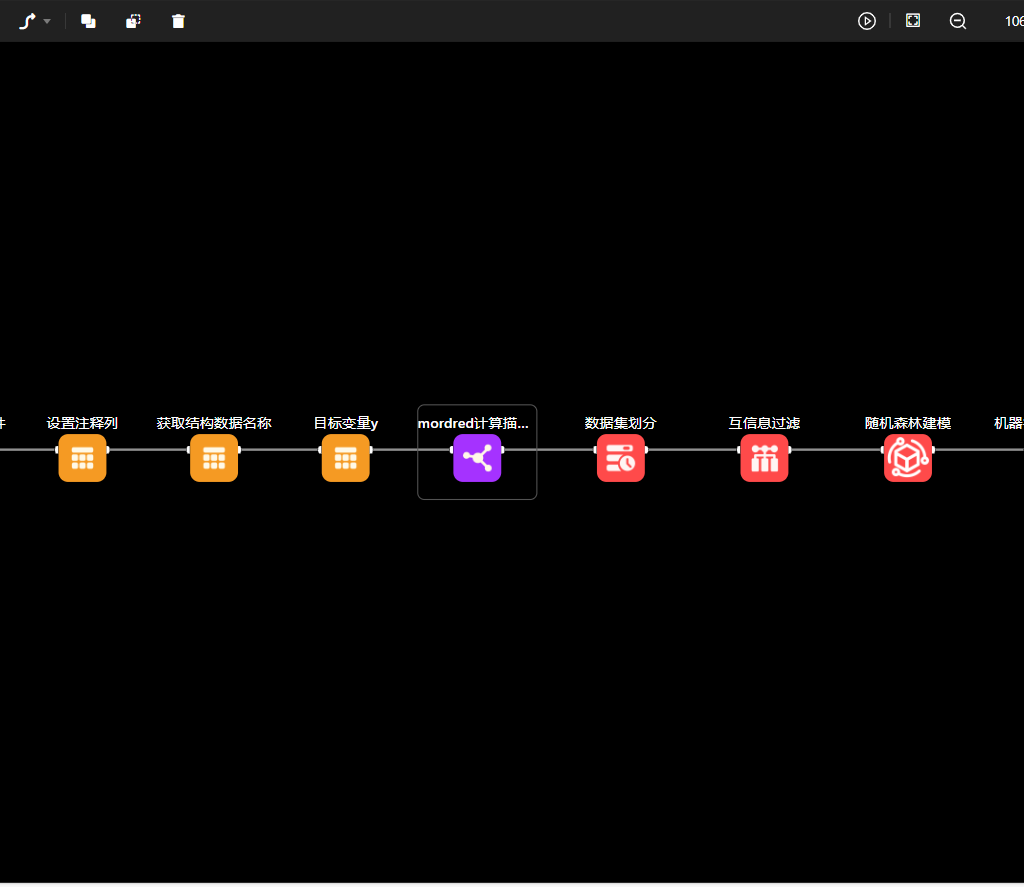
One of the standout features of MaXFlow is its “Crystal and Molecular” visualization interface. This powerful tool allows users to construct and visually edit material microstructures online, supporting a diverse array of structures, including small molecules, polymers, crystals, and more. By providing a user-friendly platform for creating molecular models, MaXFlow directly addresses the question of what would be the advantage of using a molecular model. Researchers can easily manipulate and visualize microstructures, leading to more accurate simulations and better insights into material behavior.
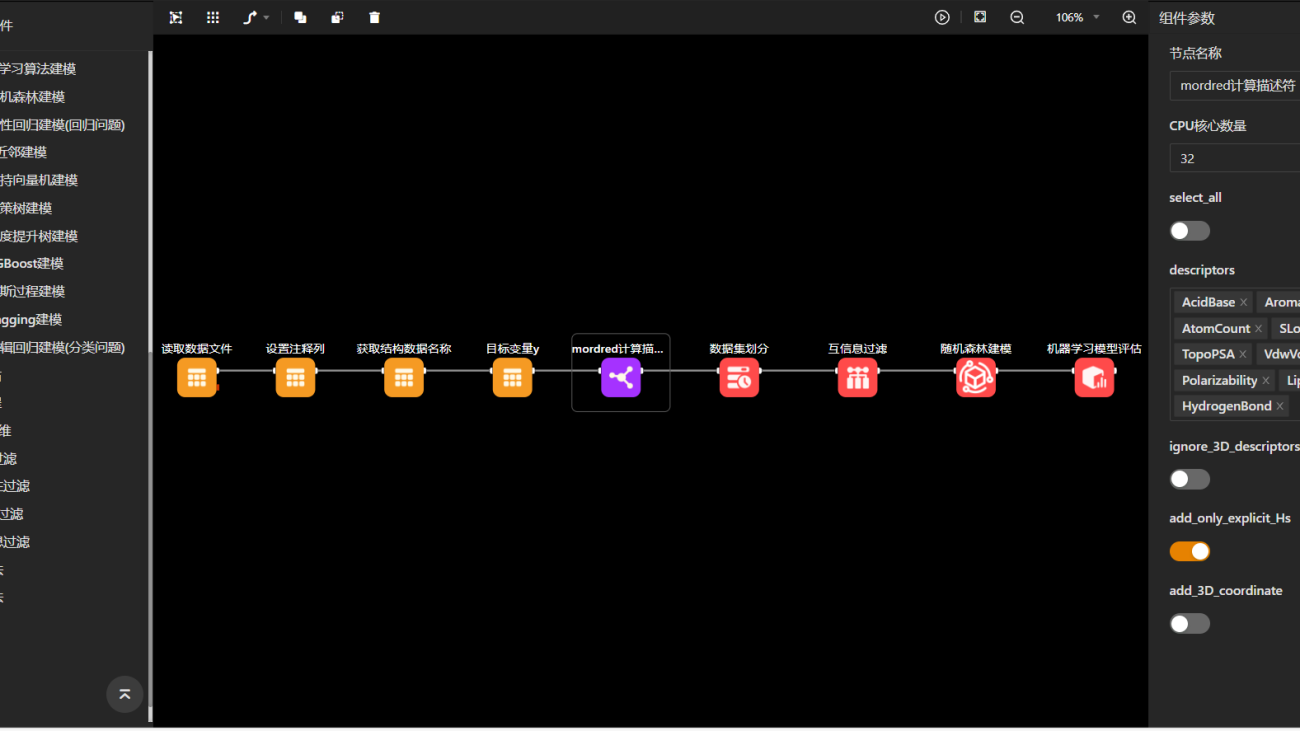
Furthermore, MaXFlow supports advanced complex microstructure modeling, enabling the creation of cross-linked structures based on defined reaction mechanisms. This capability allows users to generate models with varying degrees of cross-linking, which is essential for studying complex materials. By using molecular models, researchers can explore how different structural configurations affect the properties of materials, gaining a deeper understanding that can facilitate the development of new applications.
Conclusion
In summary, understanding what would be the advantage of using a molecular model is crucial for advancing research in material science. MaXFlow provides the tools necessary to create sophisticated molecular models, enabling researchers to predict material behavior accurately and innovate effectively. The platform’s capabilities in microstructure design and complex modeling further highlight its significance in the field.
For organizations looking to enhance their material science research, we highly recommend NeoTrident. With its commitment to leveraging advanced simulations and AI, NeoTrident is poised to support your efforts in material design and development, helping you achieve your R&D goals.